Contents
Everything you need to know about Advances in Cement Clinkering
[wpecpp name=”package” price=”75″ align=”center”]
by F. P. Glasser*
Portland cement is a manufactured product made by sintering of appropriate oxide components in the presence of a reactive liquid. A large technical literature exists on the subject. For example, the operation of the rotary kiln itself is described by Peray (1986) while chapters of Lea’s Chemistry of Cement and Concrete (Hewlitt,1988) give specialized background. Figure 3.4.1 shows aschematic of cement raw meal transformation to typical clinker products as a function of temperature during cement manufacturing.
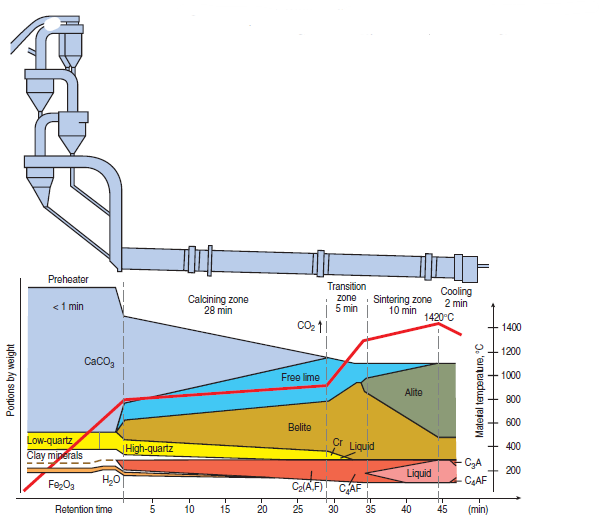
Figure 3.4.1. Transformation of raw meal to clinker products during cement manufacturing. The red line (right hand scale) shows the temperature profile.
The batch feed, often termed ‘meal’ or ‘raw meal,’ comprises mainly four oxides: CaO, Al2O3,Fe2O3 and SiO2.These oxides may be present in raw materials as various minerals: for example, CaO is typically present as CaCO3, calcite, while alumina and silica may be present as one or more complex minerals, e.g., kaolinite and other clays. Other components such as MgO, TiO2,Na2O, K2O and SO3,are generally present at the 0.1-5% level although, as will be shown,good chemical reasons exist for placing upper limits on the maximum permissible contents of several of these oxides. Figure 3.4.2 shows the material flow in the rotary kiln.
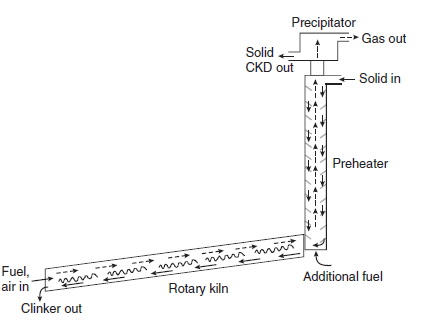
Figure 3.4.2. Raw material flow in a modern kiln. The slope of the rotary kiln is exaggerated. Abbreviation: CKD is cement kiln dust.
The rotary kiln, coupled with a suspension preheater, is thermally the most efficient processing method for conversion of raw meal to clinker. The raw materials intended for clinkering are blended and ground before being introduced into the preheater. In the preheater, water and CO2 are expelled from the batched raw materials; calcined feed emerging from the preheater is discharged, typically at 800°-900°C, directly into an inclined rotary kiln. As material flows slowly downslope in the rotary section of the kiln, temperatures reach a maximum of ~1450°C: perhaps more when dust-like particles remain suspended directly in the flame. Combustion conditions can be achieved with almost any hydrocarbon fuel: worldwide, coal is the preferred fuel but, depending on cost and availability, gas or oil can also be used. Much interest exists in using combustible waste as fuel: rubber tires, paper, wood, used solvents and lubricants, agricultural residues, etc., have all been used successfully in the kiln to supplement or replace conventional fuels.
Since the mineral components of the combustion ash mostly enter the cement clinker product, the composition and amount of ash derived from fuel have to be factored into calculating the raw material balances. The long dwell time of combustion gases in the combustion zone of a cement kiln, typically several seconds, may be compared to commercial incinerators which give only a few hundred milliseconds residence time. This long dwell time, coupled with oxidizing conditions, makes the kiln a very efficient combustor of wastes.
Historically, cement kilns have utilized a wet or semi-dry feed because wet grinding of the raw meal was used. The wet process requires a long rotary kiln and temperatures of the raw meal increase slowly, particularly during drying: evaporation of water occurring at 100°C is endother-mic as is decarbonation, the latter occurring in the range 600-900°C. After drying, the raw meal is successively dehydroxylated and decarbonated prior to formation at high temperatures of the main clinker phases. However, advances in grinding technology, including dust control, mean that dry grinding is now competitive with wet processes. While many wet and semi-dry kilns continue in use the greater thermal efficiency of the dry process makes it the preferred system for new installa-tions. Arrangements for fuel and gas flows are however relatively complex in the dry process owing partly to the need to introduce additional thermal energy into the hot zone of the preheater but the overall improvements in thermal efficiency amply justify the added technical complexities of managing fuel and air.
Finished clinker is usually rapidly cooled and is finally interground with other materials. Tradi-tionally gypsum, CaSO4·2H2O, or some other reactive calcium sulfate mineral (hemihydrate or active anhydrite) is added. The purpose of adding calcium sulfate in somewhat soluble form is to retard the initial set of cement and thereby ensure sufficient workability time. Sulfate is a cheap and efficient retarder while at the same time it does not significantly reduce the final strengths. However, many modern codes also permit addition of other mineral, pozzolanic, or reactive substances to cement: ground limestone, fly ash, blast furnace slag, silica fume, metakaolin, and natural pozzolan are interground with clinker and gypsum during finishing.
SURVEY OF REACTIONS OCCURRING DURING CLINKERING
The main reactions occurring during clinkering involve changes in chemistry owing to 1) loss of material from the batch, e.g., elimination of H2O and CO2 from the solids during calcination, 2) gains of material from the non-combustible ash components of fuel, and 3) mineralogical changes induced by pyroprocessing. Minerals which develop in response to the high temperature are those which are stable at high temperatures and also reflect the altered chemical balances result-ing from expulsion of volatiles such as H2O and CO2. The minerals which develop are, with few exceptions, subject to the additional restriction that they must be stable when in contact with melt.
Since the high-temperature portion of the clinker process closely approaches a thermodynamic equilibrium, processes occurring during heating can most appropriately be described in terms of phase equilibria and thermodynamics and of the reaction kinetics appropriate to achieving equi-librium. Experience teaches us that certain departures from equilibrium may occur, especially during clinker cooling, and if we are to understand the relationships between composition, miner-alogy, and properties of finished clinker, these departures and their impacts must be explained and their overall influence on the clinkering process calculated. Since these departures from equilib-rium are kinetically controlled, approaches based purely on equilibrium thermodynamics need to be modified and extended to embrace the relevant kinetic aspects; fortunately, the number of modifications needed are relatively few and their chemical and mineralogical consequences are often reasonably well characterized as a result of practical experience: for example, of clinker microscopy. Examples will be given subsequently.
During the heating process, considerable reaction occurs within raw meal at subsolidus tempera-tures. Broadly, these reactions can be divided into four categories, depending on the mineralogical nature of the products obtained. Either the early-formed products may persist into the final clinker, e.g., belite (C2S), or they may have only a transient existence. An example of the latter is the formation of spurrite, 2Ca2SiO4·CaCO3, formed by reaction at ~800°C between CaCO3 and silica; it subsequently decomposes to Ca2SiO4 and CaO as temperatures rise above ~850°-900°C. A third type of product concerns phases which form on rising temperature, are resorbed when melt-ing occurs, but reappear during cooling; ferrite, Ca2(Fe,Al)2O5, is an example. Finally, some phases formed during the rising temperature portion, while thermodynamically unstable at peak clinker-ing temperatures, fail to react and persist through the rest of the clinkering process. MgO is an example: while up to 6-8 wt. % fine-grained MgO, periclase, can in theory dissolve in the clinker melt phase, MgO may become “dead burnt” such that it becomes resistant kinetically to dissolution and fails to dissolve in the clinker melt with the result that free periclase persists into the final product.
This persistence is undesirable and is a potential cause of delayed expansion in hardened concrete. However, with few exceptions, such as MgO, there does not appear to be any significant body of evidence showing that undigested material persists in properly ground clinker and, moreover that phases formed in the intermediate stages of reaction significantly influence the final properties of the product; the high temperature portion of the clinkering cycle tends to erase most influences arising from mineral formation during the precursor stages. Thus with the exception of oversize material which fails to equilibrate with melt, e.g., large quartz grains, free lime and periclase, we are largely free to concentrate on processes occurring in the hot zone and during subsequent clinker cooling.
The high temperature equilibrium state is characterized by the coexistence of two silicate phases, Ca3SiO5 and Ca2SiO4,with melt. These solid phase compositions are idealized: much minor element substitution occurs, e.g, of Al for Si and of Mg for Ca, with the result that solid solutions based on these idealized formulae occur. Moreover, even micron-sized crystals of a particular phase may be compositionally zoned. To avoid attributing a specific chemistry to the solid phases of clinker, they are often referred to by name designations rather than formulae. For example, ‘alite’ and ‘belite’ are used to describe solid solutions based on Ca3SiO5 and Ca2SiO4 respectively. As will be shown, the changing chemistry affects the polymorphism of the phases; polymorphs differ in crystalline structure and, owing to cooling history, in internal microstructures, hence the terms alite and belite usefully embrace a range of polymorphic variants, microstructures, and chemistries. A few clinker compositions, especially those rich in Al or Fe, may contain a third crystalline phase at clinkering temperatures, either Ca3Al2O6 or Ca2(Fe,Al)2O5,but this is unusual: most, if not all, of the aluminate and aluminoferrite phases form by crystallization of melt during clinker cooling.
The detailed series of mineralogical changes occurring in the kiln have been known in outline for many decades. While some reaction leading to formation of belite occurs at temperatures as low as 900°C, the bulk of reaction leading to formation of the main clinker phases occurs in the presence of melt, above ~1330°C. The solid state reactions occurring during early stages of heating of the raw material have long been a source of fascination and challenge to investigators: “challenge” because it is difficult directly to sample the contents of a rotary kiln while it is in operation. Many investigations of the reactions occurring during intermediate stages of clinkering have therefore utilized shutdown periods to sample partially-burnt clinker as well as material accumulated on the refractory walls of the kiln. However, material removed from the kiln walls has often experienced abnormally long dwell periods at elevated temperatures and may have concentrated trace compo-nents in the batch, e.g., P2O5, and F. Although the chemistry and mineralogy of these products is interesting, it may not be relevant to the freely-moving calcine, which comprises the largest and most representative portion of the clinker, but which also passes through this stage relatively rapidly. The vapor phase also transports material within the kiln: a complex sulfur cycle occurs which involves, in part, transport through the vapor phase. This cycle is discussed subsequently and it is shown that the vapor pathways constitute the least-known part of the clinkering cycle.
Most of the desirable silicate phases in clinker, alite and belite, form above 1338°C in the presence of melt. Figure 3.4.3 shows schematically the situation which obtains during the initial formation of melt. Much unreacted but decarbonated raw material remains at this stage: the figure shows repre-sentative grains of CaO and SiO2.These dissolve in the melt and once melt is created in the regime of rising temperatures, these components – mainly CaO and SiO2 re-precipitate as the crystalline miner-als stable at >1330°C. The melt wets both the reactants and the growing crystals strongly with the result that the permeating and reactive liquid phase transports matter from reactants to products. Product nucleation and growth rates balance, such that crystals of the stable product phases nucleate and grow to 5-50µm within the normal dwell time in the hot zone, 30-60 minutes.
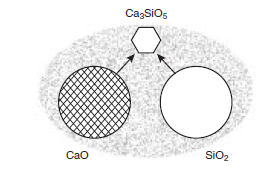
Figure 3.4.3. Schematic, showing how CaO and SiO2 react at ~1450°C with formation of products, Ca2SiO4 (belite) and Ca3SiO5 (alite) (only the latter is shown) by dissolution, transport through the melt and precipita-tion of the stable phases. The relative proportions of lime and silica are typically close to 3; the amount of melt (stippled) is exaggerated.
Conditions at this point have a major influence on clinker quality. It is important that:
• The CaO content of the batch is high, but not so high as to leave an excess of lime which cannot in theory be dissolved and combined as alite or belite: the “lime saturation factor,” defined in Appendix 1, or a Bogue calculation are often used as simple measures of saturation.
• A satisfactory compromise is achieved between grinding the reactants, especially to reduce the content of physically larger grains which are difficult to dissolve, burning time, maximum temperature attained, time in the hot zone and kiln load.
• The raw mix constituents have been adequately dispersed: agglomerates consisting of small grains of one mineral, e.g., quartz, must be avoided because monomineralic agglomerates tend to behave like a single large grain with respect to ease of dissolution. While quartz or its high temperature variants, tridymite and cristobalite, are rare in clinker, belite clusters, or “nests” frequently develop as a result of silica-rich inhomogeneities which have incompletely reacted with CaO and melt. The result is signalled microscopically by locally high concentrations of belite and, in all probability, free lime in excess of that calculated from the lime saturation factor.
Since alite and belite mainly grow from melt, they have the opportunity to saturate themselves with other dissolved species present in the reactive melt. The melt contains both major and minor constituents, including alkali, magnesium, iron, and aluminum; partition of these elements, coupled with phase transformations occurring during cooling, gives rise to the characteristic microscopic textures of clinkers, which are preserved throughout subsequent stages. Typically a cooled clinker exhibits angular, pseudohexagonal alite crystals and slightly more rounded and smaller belite, frequently twinned on a microscopic scale. Both alite and belite are embedded in a fine-grained matrix, or groundmass, developed by crystallization of former melt. Campbell (1999) offers many optical micrographs which illustrate the range of microstructures in normal and abnormal production.
The final stage, of clinker grinding, is accompanied by addition of other materials: for example gypsum. In recent years, the amount and range of additions have increased to include fly ash, slag, calcined clay and natural pozzolanic materials. These are of course present as separate granules or agglomerates and their texture and distribution within ground clinker have an important influence on subsequent hydration. Organic “grinding aids,” usually in the form of water soluble organics, e.g., amines, are sometimes added at this stage. Coupled with other organics, traces of lubricants and organic materials in gypsum, finished cements have a perceptible organic content.
SURVEY OF REACTIONS OCCURRING DURING CLINKER COOLING
During clinker cooling, the microstructural relationships developed at peak temperatures are broadly preserved. Thus alite, which is thermodynamically unstable below ~1275°C, is preserved almost quantitatively during normal cooling. However a series of subsequent reactions occur during cooling which affect the detailed phase composition and form secondary microstructures which are superimposed on the primary microstructure.
An important principle concerning the nature and sequence of cooling reactions is that reaction occurring between phases in the course of cooling is limited by kinetics: mass transport across phase boundaries is typically slow. Because of the rapidity of cooling, phase boundaries developed at or near peak clinkering temperatures tend to be preserved. Thus at clinkering temperatures, the clinker itself (neglecting pores) consists of a melt which permeates and envelops alite and belite crystals: the crystals, which comprise ca 70% of the clinker, have atomically sharp interfaces with melt, and a discontinuity in chemical composition must therefore occur at crystal-melt interfaces. These interfaces persist to a large extent without change during the cooling process. In other words, the alite, belite and melt undergo changes essentially independently from each other and we can, a few exceptions apart, discuss the cooling behavior of each portion of the clinker mass – alite, belite and melt – separately from others. As a consequence of these restrictions, reactions occur-ring during cooling are not simply the reverse of those encountered during heating and much of the technology of modern portland cement derives from a detailed understanding of the cooling reactions and the ability, at least in part, to control them. In reviewing these changes, emphasis will be placed on those phenomena which have, or are believed to have, an important influence on clinker properties.
Cement clinkers are usually cooled relatively rapidly, at least in the temperature range down tov~1000°C, for three reasons; firstly, to facilitate heat recovery from hot clinker; secondly, because the alite phase might dissociate if cooled slowly and, finally, because of the physical difficulties of transporting and storing large masses of partially molten or extremely hot clinker. Thus the rela-tively rapid cooling experienced by clinker has important consequences to the microstructure, mineralogy, and phase composition of the product and supports the contention that cooling occurs without significant mass transport between phases.
The alite phase is unstable below ~1275°C, decomposing to mixtures of Ca2SiO4 (belite) and CaO. Fortunately the decomposition kinetics are sufficiently slow as to preclude significant loss of alite during normal cooling. Mohan and Glasser (1977-a,b,c) studied the decomposition of both chemi-cally pure Ca3SiO5 and alite, the latter doped with Al, Fe, Mg, and Na. A rate maximum for alite decomposition occurs at ~1050°C but even at this maximum, the lengthy induction period preced-ing significant (> 1%) decomposition ensures that decomposition occurring during normal clinker cooling is negligible. However, two conditions were highlighted which could potentially initiate alite decomposition; cooling in an atmosphere rich in water vapor and the presence of molten sulfates. Figure 3.4.4 shows decomposition data, as inferred from the increase in free lime content, as a func-tion of water vapor pressure at 1055°C. Although the time to achieve a given fraction of alite decom-posed, shown in Figure 3.4.5, is significantly reduced in water-rich atmospheres relative to a dry atmosphere, the acceleration is not believed to be sufficient to produce technical problems in normal production. However, the presence of molten alkali sulfates induced rapid decomposition.
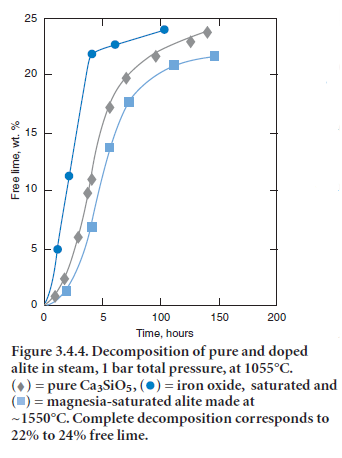
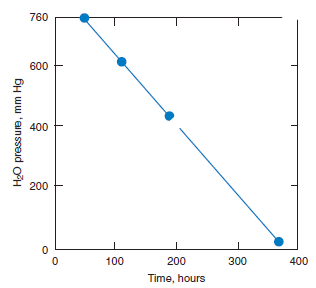
Figure 3.4.5. Time taken to reach 50% alite decomposition as a function of H2O pressure at 1055°C.
Since molten sulfates are likely to be sparsely distributed in real clinkers, it is believed that, at worst, decomposition of alite might be confined locally to alite crystals which were in physical contact with molten sulfates condensed onto or occluded within clinkers.
As clinker cools, the melt phase crystallizes. Preser-vation of melt as glass is uncommon in portland cement, although glass has occasionally been reported to occur at ambient in white portland clinkers: melts low in iron oxide and enriched in alkalis are better glass formers than the iron oxide-rich melt in portland cement. Since the freezing reactions are exothermic, substantial heat transfer must occur at interfaces between melt and growing crystals to dissipate the heat of crystallization; as a result, strong thermal gradients develop even within relatively small clinker grains. Of course these local gradients are superim-posed on other gradients introduced by rapid cooling. Thus thermal gradients occur on both macroscopic and microscopic length scales in cooling clinker. Figure 3.4.6 shows the microscopic gradients which arise in the vicinity of a crystal growing from melt.
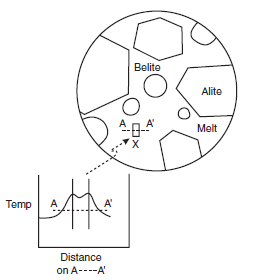
Figure 3.4.6. Local temperature gradients arising in an undercooled clinker. A crystal of an interstitial phase, X indicated by a small rectangle, nucleates and grows with release of heat. As a result, a local thermal gradient arises, shown schematically in the inset. Physical boundaries of the growing crystal are shown by two parallel lines in the inset.
The macroscopic gradient follows from the general relationships governing the thermal conductiv-ity of the sample and the heat capacity of the solids in response to cooling conditions while the second, microscopic, gradient shown in Figure 3.4.6 results from the attempt of individual crystals to continue growth while at the same time dissipating their heat of crystallization. Release of heat of crystallization at crystal-melt interfaces relieves the undercooling locally and raises the tempera-ture of the melt adjacent to the growth surface closer to that of the true liquidus. As the liquidus temperature is approached, growth slows as a consequence of self-heating. Moreover the condi-tions for continued crystal growth are additionally complicated by accumulation of chemical gradients at interfaces: the growing crystal will not in general have the same composition as the melt, with the result that atom species rejected by the crystal concentrate in the melt phase adja-cent to the interface.
During clinker cooling, the rejected species cannot diffuse away with sufficient rapidity to permit continued rapid crystallization. In extreme cases, this may lead to the liquid becoming locally satu-rated with respect to another crystalline phase(s). One expression of this condition is that the surfaces of the growing crystal become “decorated” with minute crystallites of a supernumerary phase or phases. These decorations are subsequently occluded by the growing crystal which, when examined subsequently in cross-section by optical microscopy, appears to contain “phantom” crys-tals, the geometrically regular outlines of which are revealed by numerous small crystallites of an exotic phase. Thus the phantom matrix crystals also tend to be compositionally zoned, as will be explained subsequently, and are additionally decorated by crystallization and occlusion of second-ary phases.
These restraints upon growth mechanisms often operate differentially over the length of a crystal, with the result that crystals forming in undercooled melts tend initially to grow with elongated morphologies: the direction of elongation typically extends parallel or sub-parallel to the local generalized thermal gradient.
However, the prevailing general thermal gradient,upon which other gradients are superimposed by crystallization and release of heat, coupled with other factors such as diffusion of species in the melt, may also result in crystallization becoming favorable in remote regions distant from the immediate vicinity of the contact between growing crystal and melt. This allows another mecha-nism of growth to occur when strong undercooling of the melt obtains; crystals develop multiple branches, partly in order to rapidly access regions of liquid which have a higher concentration of nutrient species and partly to access regions of undercooled melt not affected by local heat release. Such crystal morphologies are termed ‘dendritic’ and are characteristic of melts which have crystal-lized with undercooling but which also have low nucleation densities: the ferrite phase frequently grows under these conditions. Polished and thin sections of commercial cement clinker typically reveal two-dimensional slices through arrays of sub-parallel ferrite dendrites – see Campbell (1999) for examples. In extreme cases, dendritic growth is unable to accommodate the thermal and compositional gradients with sufficient rapidity, with the result that new nuclei develop spontaneously in the remaining bulk melt. These considerations result in the generally fine-grained nature of the interstial phases and explain the range of complex morphologies which are observed, even within a single clinker lump. These crystallization phenomena are strikingly similar to those occurring in other supercooled liquid systems, e.g., of metals, alloys, and slags, and give confidence in the interpretations.
The partition of elements between crystalline phases and melt is dependent on crystal structure and temperature. Considering an individual phase, e.g., ferrite, the partition of major components such as Al and Fe is temperature dependent. The relevant phase data are relatively well known, from which it would be predicted that the maximum extent of Fe enrichment would occur in early-formed crystals. Thus it is commonly observed that as crystallization proceeds, the first-formed ferrite is Fe rich. Successive accretion to individual crystals thus frequently results in ferrite with iron-rich cores, the Fe/Al ratio of which decreases in outer layers.
However, the presumption of a temperature-dependent equilibrium between melt and crystal may not be maintained: with rapid cooling, crystallization may occur with such strong undercooling that equilibrium is no longer attained. Cement clinkers reflect both sets of conditions, so it is also important to examine metastable crystallization with strong undercooling and its impact on element distribution.
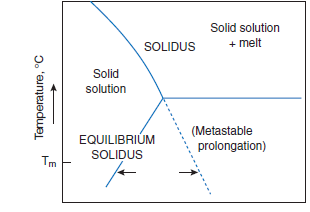
Figure 3.4.7. Illustrative diagram, showing how with decreasing temperature, solid solution may increase as a consequence of metastable crystallization. Arrows for metastable crystallization at a selected temperature, Tm,delineate differences in composition, the extent of which will generally increase as Tm is decreased.
Crystal growth in the metastable region permits the attainment of compositions which would not be stable under equilibrium growth condi-tions: this is discussed subsequently with respect to belite. Figure 3.4.7 illustrates the general prin-ciple. For typical impurities in a particular phase, e.g., alite or belite, the maximum extent of solid solution is often encountered at the solidus temperature. If equilibrium is main-tained during continued cooling, solid solution decreases in the subsolidus range. However, with non-equilibrium cooling the composition of the solid need not follow the stable curve but may instead follow a metastable prolongation of the solidus curve. Newly-formed solids which crystallize under these conditions, such as the aluminate phase, tend to have compositions lying on the metastable prolongation of the solidus rather than on the equilibrium curve.
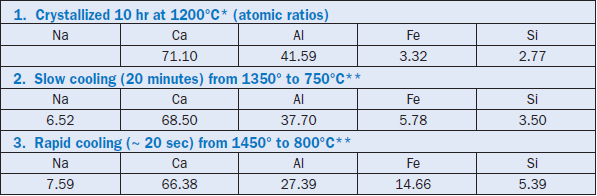
Although the example given is for a two-component system, the same principle is observed to operate in chemically more complex systems of three or more components. As a practical example, Table 3.4.1 contrasts some C3A compositions, some stable and metastable, achieved by crystalliza-tion with undercooling; the data are taken from Han and others (1981). When the rapidly cooled composition is compared with those of C3A crystallized under equilibrium conditions, it is seen that the extent of solid solution is much enhanced:crystallization occurs along the dotted line of compositions shown in Figure 3.4.7 and consequently much exceeds the maximum solid solution encountered at equilibrium. The slowly cooled composition is probably intermediate between fast cooling and the equilibrium composition, as indicated by its intermediate Al and Fe contents.
Table 3.4.1. Some Stable and Metastable Compositions of the C3A Phase
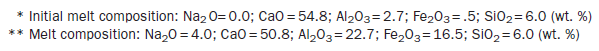
The phase composition and microstructure of clinker are influenced by the nature of the clinker batch and the clinker composition. These changes may be followed with the aid of Figure 3.4.8.
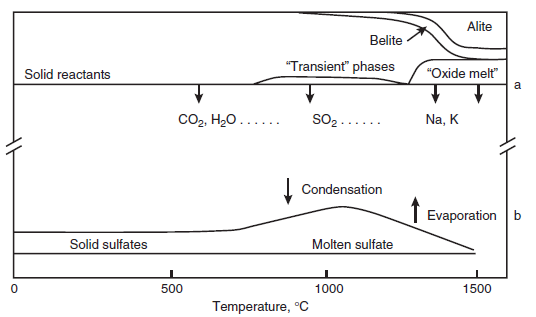
Figure 3.4.8. Schematic showing phase development in clinker as a function of temperature. The “oxide” portion is shown in “a,” the sulfate portion in “b.” Vertical arrows indicate vapor transport processes responsible for mass transfer between (a and (b).
During initial heating, water and CO2 are expelled. Many phases having a transient existence may develop. For example, lime and various calcium aluminates form transient solid phases such as the mixed carbonate-silicate phase scawtite. But as peak clinkering temperatures are approached, phases with upper limits of thermal stability, with low melting points or high vapor pressures, or some combination of these, largely vanish. Some belite, ferrite, and aluminate form, e.g., C3A, but at temperatures above 1338°C, extensive partial melting occurs and much or all of the aluminate and aluminoferrites melt. At and above this temperature, alite becomes stable and develops rapidly as transport through the melt phase facilitates its crystallization. At peak clinkering temperatures, alite, belite and melt coexist. Proper attention to avoiding oversize materials in raw meal should thus enable all quartz and lime to be assimilated in the burning zone. The growing alite and belite crystals may mechanically occlude other phases, solid or melt, including patches of molten sulfates.
During cooling, metastable phase compositions may be achieved especially amongst phases which crystallize from undercooled melts: ferrite and aluminate are the most important examples. Not surprisingly, these processes affect the amount of phases which are present in the final clinkered product. Clinker cooling thus is not simply the reverse of heating: for example, it is essential that alite formed at high temperature should be preserved during cooling even though it is metastable below 1275°C, at or below which temperature it disproportionates to lime and belite. The melt phase crystallizes during cooling, often with strong undercooling, giving rise to a fine grained dendritic structure, mainly consisting of ferrite and C3A phases. The rapid cooling enables novel dendritic microstructures to develop and solid solution compositions obtained in the course of crystallization with undercooling may exceed the limits of substitution achieved under equilibrium conditions. A series of polymorphic transitions in alite and belite occur, affecting both the crystal structures, microtexture, and reactivity of the main clinker phases: those affecting belite will be discussed subsequently.
TRANSPORT MECHANISMS IN THE KILN: THE ALKALI AND THE SULFUR CYCLES
Bhatty (1995) has recently reviewed the role of minor elements in cement manufacture and use. Many of the minor elements are essentially non-volatile and their impact on clinker properties is reviewed. In this presentation I therefore concentrate on volatile components, about which less information exists.
The most important mass flows in the clinkering cycle are the counter-current flows of solid and of combustion gases. Owing to the presence of volatile components, internal transport cycles within the kiln are of considerable importance to mass transport and influence clinker quality and kiln operation. The most important constituents in respect of internal transport cycles are chlo-rine, fluorine, sulfur, sodium, and potassium. However in modern practice, chlorine in raw meal and fuel is kept low, although other volatile components may be relatively abundant – especially sulfate and alkali – and participate in a cycle of evaporation and condensation. Sulfur and alkalis are often associated in one such complex transport cycle. The intention of the present review is to supplement Bhatty’s review (1995), to provide an integrated approach to understanding the flow of sulfur through the kiln and relate it to the alkali cycle.
Sulfate is introduced into the kiln in fuel and from raw materials. It may be introduced as sulfate, e.g., calcium sulfates in raw meal, or in chemically-reduced form in minerals, e.g., pyrite, in organosul-fides, or as constituents of oil, tar or “sour” gas fuels. The relative importance of the various sulfur speciations input to the kiln thus depends on the compositions of the raw materials and fuels. However, all the sulfur introduced into the process must eventually appear in one of three output streams: in clinker, cement kiln dust (CKD), or as gaseous emissions, with the additional but undesir-able possibility that some may be temporarily retained in the kiln as annular deposits, termed ‘kiln rings.’ The balance between outputs depends quite crucially on the inputs as well as the particular process being used: wet, dry, semi-wet, or, if the dry process is being used, on temperatures in the suspension preheater, temperature gradients within various parts of the operation and the amount of bleed air. It is probably true that as fuel economy becomes more important and, as a consequence, air is kept nearer the minimum requirements, the sulfate contents of clinker have increased.
Sulfur inputs are normally well known as a result of the widespread use of X-ray fluorescence for the on-line quality control of raw materials. XRF does not, however, indicate sulfur speciation. Thus sulfur may be present in the chemically-reduced sulfide state, e.g., as S22- in pyrite, where its oxidation state is S1-,or in one or more of its higher, positive oxidation states. For example, sulfur occurs as S6+ in sulfate, SO42-,while the S4+ state occurs in the vapor, as SO2.Mixtures of specia-tions in fuel and raw meal are thus not uncommon. However, most cement plants operate under oxidizing conditions with a slight excess of oxygen (0.8% to 2.0% O2) in the kiln exit gas. The continuous excess of oxygen in the combustion atmosphere normally ensures that sulfur, even if introduced in chemically-reduced form, becomes oxidized, to S4+ and S6+,or mixtures thereof. The presence of reduced sulfur species, S1-,S2-, in finished clinker is typically associated with undesirable features such as the presence of unburnt carbon, poor fuel economy and excessive sulfur in gaseous emissions. Hence sulfur, irrespective of its initial speciation, should become oxidized in the course of the normal burning cycle.
Numerous opportunities exist for the gas and solid streams to interact with each other leading to establishment of an internal sulfur cycle within the kiln, mainly concerned with oxidized sulfur because the most volatile sulfur species is SO2:SO3 is unstable at elevated temperatures. Thus the decomposition reaction for CaSO4 is best written as;
CaSO4 → CaO + SO2 + 1⁄2O2
The nature of the clinkering process, especially the presence of a suspension preheater, influences where and how contact between solid and gas can occur and the consequences. Empirical experi-ence of cement clinkering suggests that a strong association exists between the transport of alkali (sodium, potassium) and of sulfate. The alkali sulfates are known to have high vapor pressures, albeit much lower than SO2,which suggests that the place to commence examination of the trans-port processes is in the hot zone.
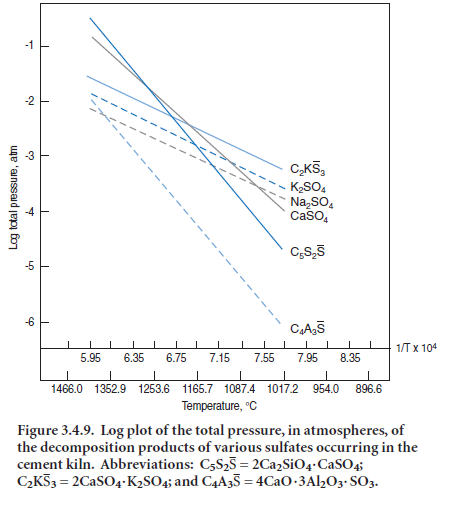
Figure 3.4.9, reproduced from data by Choi and Glasser (1988), shows the vapor pressure of some sulfates commonly associated with cement
production. Because of the wide range of vapor pressures, the data are depicted on a log pressure scale vs. 1/T, where T is temperature
in C°. The 1/T plots,while not easy to use in practical applications, do provide a nearly constant slope which is useful in data extrapolation
and mathematical modeling. Changes in the slope of the vapor pressure-temperature curves occur at phase transformations, but these are scarcely apparent on the scale of the diagram. It might be supposed that, given the importance of these substances to technology, the numerical values of vapor pres-sure of the salt-like sulfates would be well known. But this is not so: literature values scatter widely. However, the data shown in Figure 3.4.9, obtained by the Knudsen effusion method, form a self-consistent dataset. Moreover the actual numerical values generally accord with mean values reported in the literature although, as noted, literature values exhibit much scatter.
The increasing instability with rising temperature of CaSO4,C5S2Æ,and C4A3Æ is indicated by the steep slope of their vapor pressure plots. Thus these substances tend increasingly to dissociate at elevated temperatures. Since they dissociate incongruently, solid products of dissociation remain: these are CaO (from CaSO4), mixtures of CaO and Ca2SiO4 in the ratio 5:2 from C5S2Æ, and calcium aluminates from C4A3Æ. Once liberated, these solid substances are of course free to react with other clinker solids. K2SO4 and Na2SO4,on the other hand, evaporate congruently, that is, without leaving a solid residue. Since K2SO4 and Na2SO4 melt well below the temperature at which their vapor pressure becomes significant, evaporation in clinker will typically occur from a sulfate rich melt. In clinkering a lime-rich commercial cement, this liquid probably also becomes satu-rated with respect to CaO and CaSO4, although its solvent powers for CaO are poor, probably on the order of 1% to 2% by mass. The sulfate melt has been simulated in the laboratory and is found to be immiscible with the ordinary CaO-Al2O3-Fe2O3-SiO2 oxide melt by Glasser and Marr (1980); this progression is illustrated schematically in Figure 3.4.8, and the immiscibility provides justifica-tion for separate treatment of the sulfate-rich melt. Kurdowski (2002) shows how sulfates mechan-ically occluded in clinker can potentially contribute to delayed ettringite formation.
Figure 3.4.8 also shows the main clinkering reactions (top); loss of CO2 and H2O occur up to ~900°C by which they are essentially complete while the vapor pressures of SO2 and alkalis, Na2O and K2O, become appreciable only at higher temperatures. Retention of CO2,SO2,etc., at tempera-tures up to ~900°C may be responsible for the formation of transient phases in the kiln, e.g., calcium sulfosilicate and of the carbonate-silicate double salt, spurrite. However, as belite begins to form in appreciable quantities, and as temperatures continue to rise, these transient phases decom-pose. Meantime, solid alkali sulfates forming in the raw meal either by reaction or by condensation from the vapor, or both, have comparatively low melting points, in the range 600°C to 900°C. The sulfate melt is of course limited in amount by the total sulfur and alkali available in raw meal, perhaps supplemented by condensation from the vapor. Since this melt also has low solubility for aluminum and silicon, it is relatively ineffective as a mineralizer.
Once the clinker charge enters the main burning zone, melt – in this instance a CaO-Al2O3-Fe2O3-SiO2 melt – develops above ~1338°C. The “oxide” melt contains comparatively little sulfate but is a good solvent for alkali. Thus at clinkering temperatures, two immiscible liquids may occur at equi-librium: one is an oxide melt, containing virtually no sulfate, and the other is a sulfate melt in which three of the main clinker oxides, Al2O3,Fe2O3 and SiO2,are nearly insoluble. Alkalis, on the other hand, fractionate between the two immiscible liquids. Experimental studies describe this fractionation: for example, when an oxide melt having the initial composition CaO = 54.8,
Al2O3 = 22.7, Fe2O3 = 16.5, SiO2 = 6.0 was equilibrated at 1350°C with a K2SO4 Na2SO4 melt, depending on the K/Na ratio of the latter, the CaO content of the sulfate melt lay in the range 2.2%to 13.7%; Ca, furnished from the oxide melt, partially exchanged for alkali in the sulfate melt
(Glasser and Marr, 1980). On the other hand, the SO3 content of the “silicate” melt was low. Thus alkali sulfates have two pathways in the hot zone; to melt, and persist in clinker as an immiscible sulfate-rich liquid, or to evaporate. Figure 3.4.8 reflects schematically this paragenesis of melts.
Since evaporation occurs from a melt whose composition is essentially a K2SO4-Na2SO4 mixture, dissolving small amounts of CaSO4 and, as will be shown, OH1- and CO32-, the vapor pressure of melts, assuming thermodynamic ideality, lies approximately within the bounds set by the two curves of Figure 3.4.9, one for K2SO4, the other for Na2SO4.However, more quantitative treatment has been achieved by application of thermodynamic routines, as will be described.
Detailed calculations involving equilibration amongst the many species present in kiln atmos-pheres must of necessity be done by computer: fortunately the vapor species are well characterized, a good thermodynamic database exists and validated computer protocols are available with which to solve the system of equations. Barry and Glasser (2000) report results of specimen calculations for vapor-liquid equilibria for a model cement composition. In the example calculation, the overall proportions of gaseous constituents were arbitrarily taken, in kg units, as Na2O = 2, CO2 = 94, H2O = 10, O2 = 2, N2 = 117, SO2 = 1.0 and HCl = 0.2. Figure 3.4.10 shows the calculated species concentrations in the kiln atmosphere. As expected, N2,CO2,H2O and O2 are calculated to be the most abundant vapor species. However, at all temperatures of interest to the hot zone chemistry, the vapor equilibria are complex: although K2SO4 and Na2SO4 evaporate congruently, they dissociate
extensively in the vapor and, moreover, their dissociation species interact strongly with combustion product species, principally O2, CO2, CO,
H2 and H2O.
As a consequence of the dissociation of alkali sulfates and rapid reaction with other species, molecular Na2SO4 (and K2SO4) are relatively minor components of the vapor: for example, the principal sodium species are NaOH, followed by NaCl (if chloride is present), Na and Na2SO4; as temperature increases, the propor-tions of Na and Na2SO4 in the vapor increase rapidly while that of NaCl remains approximately constant with the result that at 1450°C and above the order of species concentrations, in decreasing abundance, becomes NaOH > Na ~ Na2SO4.Since the hot zone gas temperature normally exceeds that of the clinker, this calculation emphasizes the importance of a sodium transport in the vapor as two species: elemental (Na) and hydroxide (NaOH). Results of these calculations may appear to run counter to intuitive thinking, which suggests that sodium will combine strongly with oxygen. While this is true at lower temperatures, the very high temperatures encountered in the burning zone have a major impact on species distributions. Owing to chemical similarities between sodium and potassium, both elements and their speciations behave similarly, although to the author’s knowledge, detailed calculations have not been undertaken for potassium.
Results of a calculation on the composition of the “sulfate” liquid phase in equilibrium with the vapor compositions are given in Figure
1.0 3.4.11. In interpreting this figure, it is helpful to recall that the total abundance of 0.8 some species, e.g., CO2,changes little with Na2SO4 temperature whereas other species, such as 0.6 Na and SO2,have total concentrations which depend more strongly on tempera- 0.4
ture. Moreover, the movements of solid and vapor through the kiln have to be taken into account. One consequence of the countercurrent flow of solid and gas is that NaCl evaporation from semi-finished clinker tends to predominate in the hot zone while condensation tends to predominate in cooler zones having temperature 1200°C or less. Moreover, consideration of Figures 3.4.10 and 3.4.11 suggests that in condensation zones, the equilibrium melt composition will not be Na2SO4 but is more accurately described as a Na2SO4 – Na2CO3 melt: although detailed calculations have not been undertaken for potas-sium, similar conclusions are likely to emerge.A small but significant anion contribution also arises from OH1-,with the result that the melt will be strongly basic, and will attack refractories should the possibility of contact occur.
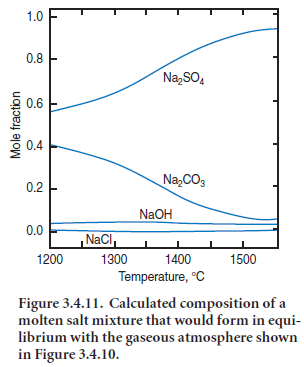
The presence of carbonate dissolved in the molten “sulfate” phase helps explain the presence of “CO2” as reported in clinker analyses, even when made on fresh clinker. The CO2 content of finished clinker is usually attributed to carbonation in storage, to minor calcium carbonate impu-rity in interground gypsum, etc. Indeed, this explanation does account for part of the CO2 found by analysis. However, at least some carbonate is introduced in the form of solid solution in the nominally “sulfate” phases. Because this CO2 is in solid solution, independent carbonate phases will typically not be identified by X-ray diffraction or optical microscopy. However one indirect clue to the presence of carbonate in solid solution is that it tends to stabilize high temperature alkali sulfate polymorphs to ambient temperature. For example, pure K2SO4 has a high tempera-ture phase which cannot normally be quenched to ambient. Nevertheless, the high K2SO4 phase is frequently encountered in clinker, where it has been kinetically stabilized to ambient temperatures by solid solution, including partial replacement of sulfate by carbonate.
Thus sulfur in the hot zone broadly follows two routes: some evaporates, while some remains in clinker. Since the total vapor pressure of alkali sulfates is likely to attain 10-2 to 10-3 atmospheres at peak clinkering temperatures, molten and solid sulfates in clinker can only be protected against evaporation by mechanical occlusion in other clinker minerals, by physical confinement within clinker agglomerates or by forming phases with low vapor pressures such as CaSO4.Campbell (1999) shows micrographs of sulfate-rich liquids suggestive of confinement within clinker. While the micrographs do not reveal unequivocally the origin of these sulfate-rich regions, they are believed to represent molten sulfates which have passed through the burning zone without having evaporated but which subsequently crystallized during clinker cooling. However, the present state of knowledge does not enable us to distinguish clinker feeds which are prone mechanically to occlude sulfates. As noted previously, liquid immiscibility prevents the sulfate melt from dissolving to any significant extent in the oxide melt phase, or conversely, of the sulfate melt dissolving signif-icant Mg, Al, Fe, or Si.
Evaporated alkalis, sulfur dioxide and oxygen exit the high temperature zone in a gas stream which is used to heat incoming raw meal. Therefore, exit gas temperatures decrease as heat is exchanged into relative cool solids. Eventually, the dew point of alkali sulfate may be reached and at this point, alkali sulfates will begin to condense. The condensation product obtained at higher temperatures,
>700°C to 800°C, is a melt but at lower temperatures condensation may occur as solid. The latter process is analogous to the direct formation of ice from water vapor and its subsequent deposition onto solid surfaces. Because the condensate may appear either as liquid or solid the term “dew point” is appropriate to mark the temperature at or below which condensation occurs.
To the author’s knowledge, no quantitative calculations other than the scoping studies reported by Barry and Glasser (2002) have been made of this process, so the following account remains quali-tative, at least for the present. The composition of the condensate will not be pure alkali sulfate but will in all probability include CO2 from the gas stream and clinker meal. At or above 700°C to 800°C, its chemistry will be essentially that of a (Na,K)2(SO4,CO3) melt containing a slight excess of (Na,K)OH. In normal production, this condensate will be swept back into the hot zone. But local accumulations of condensate, in the form of melt wetting incompletely-clinkered raw meal may cause the meal to become sticky and accumulate along kiln walls.
This phenomena, of self-agglomeration of raw meal, is sometimes encountered in semi-wet or wet process kilns and results in the formation of clinker “rings” marking the zone of active condensa-tion. Formation of rings is favored by steady-state conditions, such that vapor condensation occurs for a prolonged period of time at approximately the same point in the kiln and, moreover, at temperatures such that the condensate remains molten. The accumulating melt wets and pene-trates the still incompletely consolidated, semi-finished clinker and the resulting binding action, combined with the rotary motion of the kiln, forms the ring. Once established, the ring acts as a dam which partially interrupts the normal downslope flow of meal. Freezing of the molten sulfates, which mechanically binds matter comprising the rings, is facilitated by formation of a dam which is difficult to heat, as well as by reaction of initially molten alkali sulfates with solids present in the semi-finished clinker resulting in formation of solid minerals, such as calcium lang-beinite, K2Ca2(SO4)3; calcium sulfosilicate, C5S2Æ; and calcium sulfoaluminate, C4A3Æ:freezing can thus occur isothermally because the reaction products have relatively higher melting points than those of the initial condensate. For example, solid calcium sulfate may react with that of molten K2SO4,with complete freezing at the stoichiometric ratio 2 or more: at ratio 2.0, melt may vanish by the reaction:
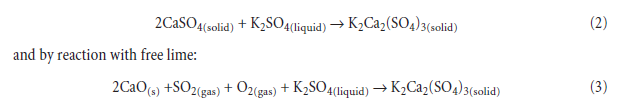
Calcium langbeinite, K2Ca2(SO4)3, has a higher melting point than that of K2SO4 or a K2SO4 –calcium langbeinite eutectic so its formation can also result in isothermal freezing. Calcium lang-beinite has no sodium equivalent, so its formation in clinker is one of the few major differences between sodium and potassium in respect of the sulfate cycle. In any case, with Na2O present, the melting point of the mixture is lower if fluxed by K2SO4.
Where the ‘dew point’ for alkali sulfates is such that condensation occurs in the vapor with forma-tion of solid phases, and if these escape deposition on clinker, they are swept further along the system until eventually removed by the precipitators: this accounts in part for the well-known enrichment of precipitator dusts in alkali sulfates.
In summary, a complex cycle of evaporation, condensation, melting and freezing of alkali and sulfates occurs in the kiln system. The coupling observed between alkali, Na and K, and sulfate is thus seen as being in part fortuitous: although Na2SO4, and K2SO4,are important in the condensa-tion process, other species not observed to condense e.g., NaOH, KOH, Na, and K are nevertheless of great importance in mediating the vapor transport processes. However, little practical use seems to have been made of the recently developed potential to model these reactions. In view of increas-ing interest in environmental management of kilns, with optimization of production, reduction of emissions, and avoidance of problems such as kiln rings and excess dust production, it is apparent that the additional tools described here can be further developed so as to incorporate the physical chemistry of the internal sulfate cycle into kiln management systems.
FLUXES AND MINERALIZERS
In the context of cement making, fluxes are classically defined as substances which facilitate reac-tion in the cement kiln; by providing a reactive liquid phase which transports matter from reac-tants to products. Al2O3 and Fe2O3 are, of course, universally used as fluxes in making portland cement: melting of CaO-Al2O3-Fe2O3-SiO2 compositions commences at or above ~1338°C and the rather fluid melt thus generated dissolves free lime and silica with precipitation of the stable phases, alite and belite, from solution. Fluxes do not, however, alter the broad thermodynamic stability of the constituent phases. Thus in the CaO-SiO2 system, Ca3SiO5 is thermodynamically unstable below about ~1275°C and the presence of the most abundant fluxes, Al2O3 and Fe2O3, does not significantly alter this relationship. However, within the thermodynamic stability range of Ca3SiO5 and at temperatures such that melt is present, the melt facilitates the dissolution of reac-tants and greatly accelerates formation of Ca3SiO5, as was depicted in Figure 3.4.3.
Mineralizers not only act as fluxes but also affect the thermodynamic stability of phases. Thus, for example, fluorine-substituted alites appear to be thermodynamically stable to as low as ~1050°. Fluorides are also powerful fluxes: for example, dissolution of small amounts of fluorine into a nominally oxide melt increases its ionicity, lowers melt viscosity, facilitates mass transport and thus accelerates formation of alite and belite under conditions favorable for crystal growth.
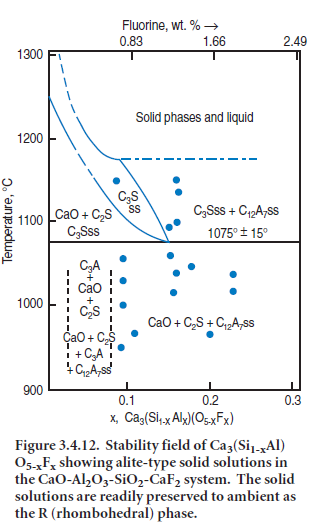
The classical distinctions between ‘fluxes’ and ‘mineralizers’ are, however, somewhat idealized. Firstly, various substances can often act in conjunction with each other to alter phase relationships more readily than either substance on its own. For example, fluorine substitutes only to a limited extent in alite where, in the absence of charge compensation mechanisms (fluorine has one negative change, but oxygen has two) the extent of fluoride substitution is limited: this, in turn, limits the scope for altering the thermodynamic stability of alite, relative to competitive phases, e.g. two-phase mixtures of lime and belite. But in the presence of both Al and F, an ener-getically-feasible charge compensation mechanism operates in alite giving rise to an extensive range of solid solutions, Ca3(Si1-x-Alx)(O5-xFx). Figure 3.4.12, after Shame and Glasser (1987) shows how solid solution can extend the thermal stability range of alite-type solid solutions to temperatures as low as ~1075°C ± 15°C. The maximum stabilizing effect of fluorine is achieved at x ~0.15, corresponding to ~1.3 wt. % F.
The multiple substitution achieved at high temperatures in the main clinker phases also affects the subsequent sequence of polymorphic inversions encountered during clinker cooling: this in turn affects clinker reactivity. For example, substitution of Na, K, Mg, Fe3+,Al, etc., into belite, achieved spontaneously at high temperatures in the course of normal clinkering, stabilizes belite as its polymorph in the course of cooling clinker to ambient temperature. Although these substituents are nominally regarded as fluxes, in their absence the much less reactive γ phase would have been obtained. Thus the classic definition of the distinction between fluxes and mineralizers tends to break down: by influencing the sequence of polymorphic transformations so as to produce a reac-tive belite phase, β, in place of the relatively inert and stable γ, these impurities – normally regarded as fluxes – can also be said to mineralize certain aspects of clinkering.
Looking objectively at the role of minor components, we need to introduce a third classification of substances: those which influence phase behavior and polymorphic change during clinker cooling, such that reactivity towards water is enhanced. Of course, the opposite behavior is also known to occur: for example, addition of borate to clinker batches. Borate dissolves preferentially in the belite phase with the result that at high lime saturation factors, alite yields decrease while clinker free lime and belite contents increase (Fletcher and Glasser, 1993).
Much of the research on fluxes and mineralizers reported in the literature has been empirical. In this context, it is noteworthy that at present i) we do not know what quantitatively are the funda-mental factors which control reactivity of selected clinker phases towards water, ii) the strategies for activation of alite and belite almost certainly differ significantly and, iii) any substances not normally present in clinkers but which are added as mineralizers may interact with fluxes already present in the batch: Al and Fe oxides are the main examples. These criteria are often not recog-nized at the outset in research programs having as their objectives the development of advanced flux/mineralizer systems.
However, it is possible to draw some general conclusions about phase development in clinkers and correlations with reactivity. For example, the enhanced reactivity of alite relative to belite arises as a consequence of differences in crystal structures of the phases. In belite, ideally Ca2SiO4,all oxygens are bonded to silicons. The silicon-oxygen-calcium bond hydrolizes in the course of reac-tion, but hydrolysis occurs slowly. On the other hand, Ca3SiO5 contains only four out of five oxygens per empirical formula bonded to silicon: its structural formula is thus Ca3(SiO4)O: the lone oxygen, not bonded to silicon, is readily hydrolized, particularly at high pH. This, coupled with the known thermodynamic absolute metastability of alite at low temperatures, results in alite being more reactive than belite. A second factor which must also be taken into account is the ther-modynamic stability of the phase under normal conditions of hydration, perhaps 0°C-100°C. For example, comparing the β and γ polymorphs of Ca2SiO4, the thermodynamic metastability of the β phase at normal hydration temperatures undoubtedly makes a substantial contribution towards its greater reactivity.
This qualitative assessment of the influence of stability and bond type on reactivity represents present understanding. However, rapid advances in molecular dynamics, which has potential to relate crystal structure and bond character to reactivity, will undoubtedly be applied in the near future to develop quantitative structure-composition-reactivity correlations.
Fluorine-mineralized systems, with promise of lowered clinkering temperatures combined with enhanced clinker alite contents, have been viewed as a promising way forward in recent decades. Interest has centered around fluorine as the mineralizer of choice: it is relatively cheap in the form of CaF2,fluorite, and is effective both as flux and mineralizer. Broadly, two strategies have been devised for its most effective use:
• Clinkering temperatures in the kiln are kept essentially unchanged from those used for normal production. In this application fluoride is used mainly to speed the clinkering process, to enable use of high lime modulii mixes, resulting in high clinker alite contents but without significant increase in the free lime contents of the resulting clinkers. Fluorine also enables preservation of rhombohedral alite to ambient, the presence of which is often taken as a favor-able indication of potential reactivity.
• Fluoride additions are used to reduce clinkering temperatures: the bulk composition is balanced such that alite contents are optimized. Moreover, the lower thermal stability limit of alite is extended to lower temperatures by solid solution. By taking advantage of the thermo-dynamic stabilization of alite to lower than normal temperatures, supposed advantages are fuel saving and less wear on kiln refractories; energy requirements for grinding may also be lowered as low temperature clinkering tends to produce a friable clinker.
Both schemes have potential advantages and disadvantages. Two technical problems exist; firstly, fluorine is itself somewhat volatile and fluorine emission, mainly as HF in gas streams, is undesir-able on account of its corrosive and toxic nature. It is a reasonable supposition that fluorine volatility will increase with temperature and on that account, high temperature clinkering is likely to be less satisfactory from the standpoint of emissions. However, the supposed beneficial impact of low temperature clinkering on emissions cannot be proved on the basis of presently available evidence: insufficient published data are available on how to optimize the process and on product properties. A second problem concerns the mineralogical distribution of fluorine retained in clinker. While it is desirable that fluorine should preferentially enter the alite phase, its actual distribution in clinker depends on temperature and on the nature of the coexisting phase assem-blage. At high temperatures, such that an oxide melt is present, fluorine partitions between crys-talline phases – alite, belite – and melt. Fluorine is not readily substituted into belite but is somewhat more readily partitioned into alite.
Fluorine is also very soluble in the melt phase and this favorable partition into the melt phase has two consequences: firstly it represents inefficient use of fluorine if the objective is to stabilize and activate alite by fluorine inclusion. Secondly, the melt phase crystallizes during cooling and the presence of even small amounts of fluorine incorporated into the melt drastically alters the miner-alogical nature of the products of crystallization. For example, C3A – in the absence of fluorine a normal crystallization product of cooled clinker – is replaced in whole or in part by C11A7·CaF2. This fluorine-substituted C12A7 type phase contains only 2.67 wt. % of fluorine, assuming the max-imum possible fluorine substitution is achieved. Hence even small amounts of fluorine are very effective in reducing or eliminating clinker C3A which is instead replaced by a C12A7·Ca(F,1⁄2O)2 solid solution. This is not necessarily a disadvantage: the fluoroaluminate reacts readily with water giving good to excellent strengths; indeed, rapid hardening cements have been developed from fluo-roaluminate – containing portland clinkers. However, such clinkers have also been associated with flash set and it is apparent that satisfactory mineralized clinkers containing the fluorinated C12A7 type phase differ significantly in properties and performance from traditional portland cement.
Another phase reported to be stabilized by fluorine is an apatite-structured phase, fluorellestadite. Ellestadite apatites, Ca5(SiO4,SO4)3(F,OH,Cl), are well known from their natural occurrences and the F end-member is readily synthesized. However, ellestadite apatite is relatively non-cementitious and behaves essentially as an inert diluent in cement clinker. Its formation in F-mineralized clinker therefore reduces the amounts of CaO and SiO2 potentially available to form alite and must be considered to represent an inefficient use of fluorine
Thus the use of fluoride-based mineralizers has both desirable and undesirable consequences: potentially desirable when calcium fluoroaluminate is used as the basis to form ultra rapid strength gain cement, but undesirable on account of its short workability time and highly exother-mic heat of hydration, which is liberated in a short period of time: also because in the presence of sulfate, much fluorine may be wasted by forming fluorellestadite and other unreactive phases.
Many of the studies reported in the literature concerning development of flux and mineralizer systems which become active during the high-temperature part of the clinkering cycle have one or more shortcomings which inhibit their practical application. It is also often difficult to obtain proof of efficaciousness. If, say, an experimental fluxed clinker is compared with normal produc-tion, what basis for comparison should be used? Is it sufficient to measure calorimetric heat of evolution in a fixed time? Or are rates of compressive strength development essential to bench-mark performance? Will specific measures of performance be adequate or is it also necessary to determine other factors, e.g., durability of the resulting concretes in service environments and inhibition of steel corrosion? Are health and safety issues associated with the particular chemical system? Finally, do unwanted but unavoidable consequences occur, as well as benefits, from the proposed mineralizer and flux combinations?
Although uncommon, mineralized cements are being produced in Denmark on a routine commer-cial basis. A few cement plants also operate successfully on indigenous, fluorine-containing raw materials. It is likely that the principal saving from the use of F would arise by reducing the thermal energy required for clinkering, but that the extent of the reduction appears to be limited. Since thermal energy is typically the cheapest of the various energy inputs to the clinkering process, develop-ments in mineralized cements appear not to have high priority, given the potential environmental problems associated with their production. On the other hand it is unlikely that the optimized phase balances have been achieved in existing processes and it is possible that a reassessment could disclose a more favorable outcome from the application of optimized combinations. Savings in grinding energy arising from a friable clinker also need to be factored into a cost-benefit analysis.
CLINKER COOLING
During clinker cooling, a series of processes and events occur which have a considerable influence on the composition of and performance of portland cement. Not only do the amounts of phases change: for example the melt present at peak clinkering temperatures freezes with production of much ferrite and aluminate, but phase transformations occurring during cooling also affect the polymorphism and reactivity of the constituent phases, especially belite, as will be shown. High processing tempera-tures also induce a significant population of defects into the crystalline structures of the phases and experience of silicate and aluminate structures discloses that on the timescale applicable for cooling clinkers, the intrinsic defect content is likely to be frozen in, or partly so.
These crystal defects may involve single atoms or ions, so-called point defects, or they may be extended defects, e.g., axial or planar defects. Extended defects are probably thermodynamically unstable in pure phases but the density of point defects is an equilibrium feature of real crystals. For example, the intrinsic defect concentration in CaO at 1500°C has been estimated at 3 x 10-9, i.e., one atom site in 3 x 10-9 (Glasser, 1998). While this is a low number, it is sufficient to facilitate diffusion in the solid and may influence reactivity during subsequent hydration. While free CaO is a minor constituent of normal clinker, similar numerical values are probably obtained for the major phases, e.g., alite. These factors – chemical substitution, defect content and type, combined with polymorphism and the sequence of polymorphic transformations, with resultant accumula-tion of strain energy – affect reactivity. Historically it has been very difficult to deconvolute these separate factors. However, much progress has been made in relating polymorphism and thermal history of belite to reactivity, as will be described.
The crystalline phases present at high temperatures undergo a series of transformations during clinker cooling. These transformations range in the extent to which structural rearrangement must occur: some solid transformations, notably those in alite, are relatively minor and probably involve only slight changes in atomic positions and bond angles and are termed ‘displacive.’ Others, notably the α’– γ transformation in belite, require the structure to be reconstituted, implying that extensive atomic migration must occur in the course of the transformation. Moreover atomic movements occurring in the course of phase transformations interact with the point defect popu-lation: these interactions are likely to introduce additional extended defects. However, this account of phase transformations in cooling clinker is simplistic; other chemically-related phenomena also occur: solid solution affects the energetics of the entire structure as well as the transformation mechanism and, therefore, the detailed sequence of phase transformations occurring during cool-ing. In fact the intrinsic defect content in most real clinkers is overwhelmed by the chemically-induced defects arising from solid solution. For example, valence-compensated substitutions in alite, e.g., of M3+ for either Ca or Si, or both, introduce extrinsic, chemically related defects and the numbers of such defects are likely vastly to exceed the intrinsic defects population. For this reason, we concentrate on the relationships between extrinsic, i.e., chemical defects, phase transformation and reactivity towards hydration.
As will be shown, phase transformations occurring on the cooling cycle are frequently accompa-nied by a decrease in the extent of stable solid solutions. Achieving a displacive phase transforma-tion in pure materials may require only short-range movement of atoms. However, in chemically more complex solid solutions, such that phase transformations require exosolution, atoms or ions may be required to diffuse over relatively long distances. Thus displacive transformations which occur rapidly and reversibly in pure materials tend to become reconstructive in solid solutions because a separate nucleation and diffusion step is required to adjust compositional differences: the nucleated phase (or phases) have to act as hosts for the excess of impurity ions. As a conse-quence, transformations which occur rapidly in pure phases may become slow in solid solutions.
The temperature-dependent sequence of inversions occurring during cooling of belite has been explained. It has long been recognized that the sequence of inversions encountered during cooling also imposes a characteristic texture on the belite phase. Based on microscopic appearance, Insley (1936) classified belites as follows:

Of these forms of belite, Types I and Ia are abundant in commercial clinkers and are therefore of greatest interest. Yamaguchi and Ono (1962) describe Type I belites as forming rounded grains, lack-ing crystal morphology, and having two or more sets of lamellae, which intersect at 60° angles. The current interpretation of the origin of these lamellae is that they arise in the course of the a→ a′inversion: the decrease in symmetry attending the inversion permits nucleation and growth of ′belite in several orientations with respect to the unique c axis of the precursor a′ phase.
Individual lamellae often exhibit polysynthetic twinning but on account of its scale, twinning is only seen at relatively high magnifications when using optical microscopy; Campbell (1999), shows a particularly coarse example of the polysynthetic twinning; in this instance, occurring in a Type II belite. The usual interpretation of the origin of polysynthetic twinning is that it arises as a consequence of the a′→βinversion, partly as a stress-release mechanism for the structural misfit between host matrix (a′) and the growing product (β). In this connection it should be recalled that solid solution introduces a two-phase region of coexisting a and a′ or of a′ and β solid solu-tions so that as temperatures decrease the proportions of the low temperature phase, either a′ or β, gradually increases. The higher temperature a → a′ inversion similarly produces twin lamellae with the result that a single crystal grain of belite which has undergone slow cooling through the sequence: a → a′→βmay contain as many as six sets of interpenetrating lamellae (Yamaguchi and Ono, 1962; Maki, 1994). The close spacing and multiple repetitive nature of the lamellar struc-tures thus developed facilitates phase transformations but, as temperatures progressively decrease below the temperature range of the relevant transformation, misfit between dilational properties of the intergrown phase or phases results in the accumulation at ambient temperatures of signifi-cant elastic strain energy which, in turn, enhances reactivity during subsequent hydration.
The distribution of chemical impurity in belite achieved during clinkering may be also rather inhomogeneous. Maki (1994, 1995) reports that alkalis tend to concentrate in the cores of individ-ual belite crystallites, which make them more reactive towards etchants, while alumina and iron oxides concentrate preferentially in the outer layers.
One of the most interesting and potentially significant reactions affecting the belite phase has been the discovery and elucidation of remelting reactions. These refer to reactions involving simultane-ous phase transformation and exosolution which occur spontaneously during belite cooling. They are termed ‘remelting’ because in the course of transformation, and with decrease in temperature, a solid host crystal spontaneously develops an internal melt. This rather unusual occurrence, of melting occurring in response to a decrease in temperatures, requires further explanation.
The potential interactions between remelting, twinning, polymorphic change and reactivity have been explored in Type 1A belite crystals by Fukuda and others (1999, 2000-a,b). At clinkering temperatures such that belite coexists with melt phase, impurities partition between crystals and melt. At ~1450°C, the belite phase coexisting with melt is the polymorph; impurities (Na, K, Mg, Fe,Al, etc.) are typically more soluble in the a phase than in a′. Upon cooling, as a belite converts to a′,impurities must exsolve in order to maintain equilibrium. Moreover, although in theory phase could simply persist metastably, study of real clinkers shows that the transformation achieves, or nearly achieves, equilibrium. The transformation proceeds by a mechanism which minimizes the amount of atomic diffusion, and is illustrated by a simple but relevant example
Figure 3.4.13 from Fukuda’s work (2001) shows the situation with respect to one such impurity, Al2O3.The upper limits of stable solid solution in the belite phase follow the line m-n-o; solid solution reaches a maximum of ~3 wt. % “Ca12Al14O33” at ~1395°C. This temperature also marks the lowest temperature at which belite remains stable. Now, in order to maintain equilibrium with continued cooling ,a must invert to a′.However, alumina is much less soluble in a′ than in a, the solubility curve of which follows p-q-r. Accordingly, a large amount of alumina – calculated tinhis example as C12A7 – decreasing from 3% to 1% approximately, has to exsolve at 1395°C in order to com-plete the inversion. However at the relevant temperature, the bulk composition of the exsolved material is molten.
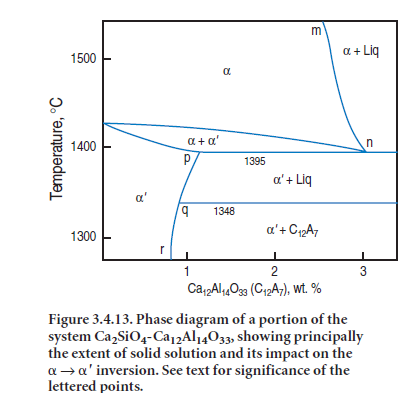
Practical observations show that at ~1395°C two processes occur during cooling. Firstly the a → a′ inversion occurs by a nucleation and growth process. The two crystal structures are coherent over the short range of inversion temperatures with the result that lamellae of ′ develop in orienta-tions controlled by the symmetry of
the precursor a phase: this minimizes interfacial surface energies between a and a′ phases . Secondly, and occurring simultaneously with the inversion, the solid exsolved material partially melts, forming local planar-shaped regions. The melt is strongly enriched in impurity (in this example, Al, but in real crystals the melt also contains alkali, Mg and Fe). In order to minimize transport distances and surface free energies, melt segregation occurs along the coherent planes of exosolution lamellae, thus accounting for the unusual geometry. In theory, this melt should diffuse out of the crystals and merge with the bulk melt but in practice, cooling is too rapid, or bulk diffusion too slow, or both, for this to occur and the melt remains trapped within individual belite grains.
With continued cooling the geometrical disposition of melt remains unchanged but it crystallizes. This initial segregation of melt on a microscopic or submicroscopic scale, followed by crystalliza-tion, affects the color and texture of belite: where the segregations are of sufficient size to be seen by optical microscopy, the resultant texture corresponds to Type 1A belite. However, in many instances belite is cooled too quickly for the segregations to crystallize on a microscopic scale, with the result that the products are not resolved on the scale of optical microscopy. The minute inclu-sions do, however, scatter visible light and the resulting scatter gives rise to optical darkening of belite: typically yellow to brown. The exosolution phenomenon thus gives rise to a range of textures and the color of belite is a sensitive indicator of the physical size of exsolved impurity regions; on the finest scale, belite remains optically clear in thin section but when viewed in plain polarized light, and as the segregations coarsen, belite turns initially yellow, then brown before visible segregation occurs on the scale of optical microscopy. Thus the color is due partly to iron in solid solution, but a more important influence is the greater scattering and absorption of blue light as a result of optical interference of the nanoscale exsolution.
As noted, the melt segregations initially developed at ~1395°C do not remain molten upon contin-ued cooling but instead crystallize. In the simplified example, the product of crystallization will be C12A7 but in real clinkers, containing a normal spectrum of oxides in solid solution with belite, ferrite phase is also produced. The optical effects resulting from exosolution of ferrite, taking into account its high refractive index relative to other exsolved phases, is much enhanced by precipita-tion of ferrite.
Fukuda and others (1999, 2000-a,b, 2001) have performed experiments to determine the impact of the remelting process on the technological properties of belites. They found that the larger the volume fraction of belite which undergoes the remelting reaction, the easier the belite is to grind and, moreover, the more reactive it becomes in the course of subsequent hydration. With continued cooling, and as the melt freezes and the resulting solids cool, increasing mismatch in the thermal dilational coefficients of the exsolved phase(s) relative to those of the host crystal creates interfacial strain. This accumulated strain is believed to be responsible for the increased ease of grinding and the subsequent enhanced reactivity of belites which have undergone the remelting reaction.
The entire cooling process, together with the features and processes which occur, is of great interest to the goal of achieving more reactive belite. Since most of the knowledge described here is of recent origin, research is still required to translate into practice the fundamental discoveries: nevertheless the results described above represent a potential major breakthrough in the systematic search for activated belites. In particular, if the remelting reaction enhances the reactivity of belite, it is neces-sary to burn the clinker at >1400°C, so that alpha belite is present at burning temperatures.
PHASE COMPOSITION OF PORTLAND CEMENT CLINKER
Cement raw meal compositions have to be formulated in terms of bulk chemistry and, because engineers are accustomed to relating the properties of a finished cement to phase composition, i.e., to clinker mineralogy, a method is required to relate raw meal chemistry, corrected for loss of volatiles and adjusted for contributions from fuel, to the mineralogy of the clinker.
Historically, chemical and mineralogical compositions were related by the Bogue calculation. Bogue was well aware of the phase relations in the CaO-Al2O3-Fe2O3-SiO2 system which had recently been elucidated by many investigators, including Bogue. He utilized a system of four linear simultaneous equations to partition the four principal oxides, CaO, Al2O3,Fe2O3 and SiO2, amongst four phases: Ca3SiO5,Ca2SiO4,Ca3Al2O6 and Ca2(Fe,Al)2O5.Free CaO, if known to be present and if determined by a separate analysis, was deducted from the bulk chemical CaO prior to solving the equations. Because subsequent steps of the calculation rest on phase equilibria in the CaO-Al2O3-Fe2O3-SiO2 system the calculation is soundly based. However, the application of the Bogue calculation ideally requires some prior knowledge of the ferrite phase composition: in the absence of such knowledge, a ferrite composition has to be assumed, e.g., C4AF. Bogue was well aware that the ferrite phase of commercial clinkers exhibited substantial variation in A/F ratios.
However, at the time, it was difficult to accurately determine ferrite phase compositions with the unsophisticated equipment available, even in state-of-the-art plants. The situation has only marginally improved with respect to the determination of ferrite composition in a particular clinker. Indeed, the ferrite is frequently zoned with respect to A/F ratio so the best that can be obtained using presently-available instrumental methods is an averaged of ferrite composition.
Numerous criticisms have been made of the Bogue calculation: potential problems with ferrite have been mentioned, but it is also noteworthy that “minor elements” e.g., Na, K, Mg, Mn, Ti, etc., are not specifically included in the Bogue calculation. Indeed, it would have been unreasonable to have done so at the time because patterns of minor element fractionations amongst clinker phases were not sufficiently well known. Possible departures from equilibrium during cooling may also affect phase compositions, although the potential impacts of non-equilibrium cooling are contentious. A more serious problem arises from recent instrumental determinations of phase content of commercial clinkers, results of which apparently show that the Bogue calculation systematically underestimates the alite content of clinkers. Two independent lines of evidence converge upon this conclusion: image analysis of clinker, as applied to determine phase composition, and quantitative X-ray analysis.
Advances in instrumental methods have enabled direct measurement of clinker mineralogy. Gutteridge (1984) pioneered quantitative X-ray diffraction and studies by Kristmann (1977, 1978) have quantified determinations by optical microscopy; a review by Kristmann (1978) summarizes the developments in optical methodologies. The X-ray methods have subsequently been much refined to take into account minor polymorphic variations in some of the clinker phases e.g., in alite. Aldridge (1982) describes the results of an interlaboratory test on the mineralogy of six clink-ers. While considerable interlaboratory variation in measured phase contents occurred, a principal conclusion was that alite contents, measured either by microscopy or X-ray, were significantly higher than calculated by the Bogue method
.
Taylor (1989) proposed revisions of the Bogue calculation to take into account recent knowledge on the distribution of minor components. Commencing with sulfate, he proposed to incorporate data on sulfate distribution amongst the minor phases as follows: the sulfate distribution into K2SO4,Na2SO4,and anhydrite (CaSO4) was made on the basis of a water solubility test; Pollitt and Brown (1968) give actual data on sulfate distributions in clinker and describe their test method which is essentially based on the rapid water solubility of alkali sulfates relative to anhydrite, CaSO4 and other Ca-containing sulfates. Alternatively, and in the absence of specific data on sulfate distributions, it was suggested that data on a set of real clinker test results be generalized and a subset of equations can be used to calculate mineral balances. Letting K = K2O, N = Na2O, C = CaO, and Æ= SO3 (all quantities are total amounts, as determined by analysis), the distribution as sulfates in moles per 100g clinker can be calculated in four steps:
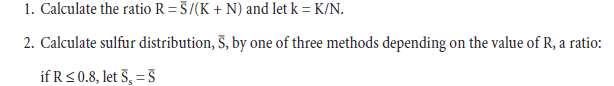

This enables sulfur to be combined and the total CaO to be reduced by the amount combined as CaSO4:application of the equations will also reduce the unassigned alkali contents.
Taylor (1989) still faced the problem, as did Bogue, of assigning the remaining four components to four phases but with the additional relaxation of assignment rules so as to admit small amounts of additional components e.g., alkali and MgO, sulfur having been treated in a preliminary step. Calculating the impact of minor components on other phase components is not so simple. Noting
Table 3.4.2. Averaged Compositions of Phases Present in Portland Clinkers (from Taylor, 1989)
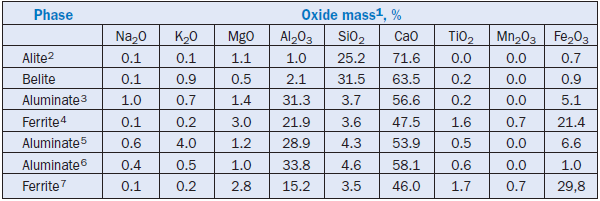
Notes
1. Data compiled by Taylor (1989), but P2O5 and SO3 values are omitted
2. Bulk clinker had 1.65% MgO and 2.8% Fe2O3
3. Cubic aluminate, typical of low alkali clinkers
4. “Normal” ferrite in clinkers with Al2O3/Fe2O3 ≥ 1.5
5. Orthorhombic C3A from high alkali clinker
6. Tentative composition of aluminate phase in white portland cement
7. Tentative composition of ferrite in a sulfate-resistant clinker
that literature studies (Yamaguchi and Takgi, 1968; Terrier, 1968) on modern cements disclose that most clinkers have weight ratios Al2O3 /Fe2O3 in the range between 1.5 and 3.0, Taylor (1989) also notes that within this range, the compositions of the principal clinker phases remain nearly constant. These compositions, taken from a compilation in of Taylor’s work (1989) are shown in Table 3.4.2. The calculation is illustrated by an example.
Step 1:
From the CaO content, deduct the CaO equivalent to CO2; also deduct any free CaO in clinker.
Step 2:
Determine insoluble residue and subtract its composition; alternatively, assume the insoluble residue is 0.3% and it is composed of SiO2 = 67% and Al2O3 = 33%.
Step 3:
Calculate sulfate balances and make appropriate corrections.
Step 4:
Correct the alite content of the clinker in accordance with its Mg content; Mg is assumed to substi-tute for CaO.
Step 5:
The analysis of the cement should preferably has been obtained before blending but if blended with gypsum or limestone, deduct any quantities either known or guessed to be present.
Step 6:
The equations to be solved are:

These results compare with a standard Bogue calculation which yields C3S = 58.6; C2S = 18.3; C3A= 9.3, and C4AF = 8.5 by mass %. Note that the Taylor (1989) calculation substantially increases the alite content with respect to the original Bogue calculation. An extended statistical trial, using data taken from the literature was offered to show that generally improved agreement between calculation and observation is obtained particularly in respect of clinker C3S contents.
Although the Taylor (1989) calculation appears to represent an improvement to the original Bogue calculation, it nevertheless has a number of disadvantages and potential inaccuracies and contains a number of empirical and arbitrary factors. Taylor himself notes that if the calculation is applied to clinkers having weight ratios Al2O3/Fe2O3 < 1.0, the calculation will give an apparently negative aluminate content: to circumvent this problem he suggests that the iron content of ferrite be arbi-trarily increased when the analysis discloses low alumina ratios. A similar situation occurs at Al2O3/Fe2O3 above 4.0, in which case a high Al ferrite composition needs to be substituted. A further restriction is that the method may not be applicable to slowly cooled clinkers or to clinkers made under reducing conditions.
The author would add other concerns: the distribution of Mg is subject to wide variations; while it may partially substitute for calcium up to a few weight %, especially in ferrite it may also appear as free periclase. This upsets the calculation unless a correction for periclase is introduced. Moreover, sulfates may appear at different stages of the clinkering cycle and may not be in equilibrium, with the result that two clinkers, each having the same nominal SO3 content, could differ significantly in sulfate mineralogy: the scheme for sulfate distribution incorporated into the Taylor calculation does not accord with equilibrium and is unlikely to be applicable to all clinkers. Moreover, the nominal “sulfate” phases may incorporate substantial carbonate, leading to systematic error. Finally, the method rests on the assumption that each phase has a fixed composition, whereas we know from microscopy and microprobe analyses that crystals are often compositionally zoned. Taylor also asserts that disequilibrium occurring in the course of clinker cooling will not by itself affect the amounts of phases formed. This is contrary to theory and conflicts with practical observations.
Perhaps a more general conclusion is that the limits of hand calculation, perhaps supplemented by computer-based computation to solve a simplified set of simultaneous equations, has been reached if not exceeded. Therefore, either the Bogue calculation should be used in its original or in various slightly modified forms, for example including a correction for free lime, or else calculations have to become more sophisticated to reflect the equilibria as well as principal departures from equilib-rium occurring during clinker cooling. To resolve these problems a fresh approach is desirable, perhaps using methods originally developed in other fields.
Great progress has been made by metallurgists in relating chemical and phase compositions. These approaches employ thermodynamics, calculating the equilibrium at each relevant temperature and tabulating cumulative changes in both the amounts of phases formed and their compositions. In this way phase compositions can be calculated either assuming equilibrium or by introducing various restrains. Numerous algorithms have been developed and quality assured for this purpose; a recent book by Saunders and Miodownik (1998) reviews such developments. Briefly, chemical species are distributed amongst phases so as to minimize the Gibbs free energy of the system. The only oper-ator inputs required are bulk composition, temperature(s) and temperature range of interest, permissible departures from equilibrium and, if relevant, pressure(s): in cement making we are mainly concerned about changing temperature and bulk composition. The database associated with the computer routine makes it unnecessary to assume a fixed composition for the individual phases: their composition(s) arise spontaneously in the course of calculations. Moreover, phase balances can be tabulated in thermal cycles in various ways, so certain well-known departures from equilibrium can also be included in the scope of calculations with less need for arbitrary assumptions.
Barry and Glasser (2002) undertook a demonstration of the method and its application to clink-ering. Calculations were performed using “MTDATA,” at the National Physical Laboratory (UK). A hypothetical portland cement composition CaO = 69, Fe2O3 = 3.6, Al2O3 = 5.4 and SiO2 = 22.0 (wt. %) was selected as the basis for calculation. Figure 3.4.14 shows the equilibrium phase distri-bution calculated as a function of temperature. Two principal restraints were introduced; firstly, that alite, once formed, was not allowed to decompose at or below its lower limit of thermal stability and secondly, that crystalline phases, once formed, were not allowed to react with melt. These restraints accord with what we know about the clinking process. During cooling, abrupt changes are predicted to occur especially in the range 1340°C to 1330°C as the clinker liquid freezes; much C3A, ferrite, and a little C2S form. What is novel is the predicted equilibrium decrease in amount of C3S which accompanies freezing. Sensitivity studies disclose that this decrease is a widespread phenomena and is not peculiar to the calculation example. However, a spontaneous decrease in the amount of a solid during cooling does not occur in practice because the rapidity of cooling in the relevant temperature range inhibits dissolution of excess C3S in melt and attainment of a new equilibrium. As has been shown, with few exceptions, each phase present at clinkering temperatures alite, belite and melt – tends to crystallize separately during clinker cooling without significant reaction with other phases. Under these conditions, the excess of C3S persists and it is primarily this phenome-non – failure to resorb C3S during cooling – which results in the underprediction of alite by the Bogue calculation.
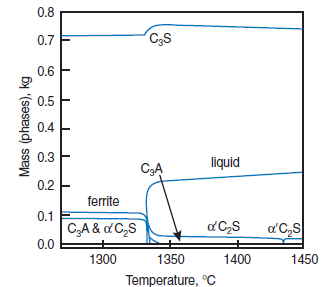
Figure 3.4.14. The masses of phases in chemical equilibrium as a function of temperature for a composition C = 0.69, F = 0.36, A=0.054, and S = 0. 22; liquid is calculated to disappear at 1334°C and as freezing occurs the amount of C3S decreases.
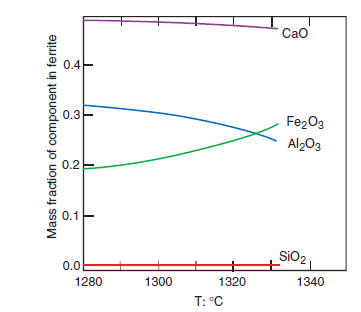
Figure 3.4.15. Calculated composition of ferrite phase with cooling of the example composition (see text). The diagram should be read from right to left to show the instanta-neous ferrite composition crystallizing from melt, commencing at -1330°C, and finishing at the freezing point, -1280°C.
This type of departure from equilibrium is well known to occur in metals as well as in nature, during the cooling of large masses of once molten or partially molten rocks. Metallurgists describe it as ‘Scheil cooling’: an accessible account is given in by Cahn and Hassen (1996). The MTDATA system is designed to accept this condition: the calcu-lation is divided into steps, each incremen-tally lowered by a fixed, user-specified temperature differential. The equilibria are recalculated at each step, typically 1°C, and the resulting mass balances tabulated and used as inputs to the next step.
For example, the composition of the ferrite phases in terms of changing Fe/Al ratio is shown in Figure 3.4.16. Note how the Fe/Al ratio decreases with decreasing tempera-ture. This results in the well-known zoning commonly observed in ferrite, in which ferrite crystals have early-formed iron-rich cores. As new layers accrete onto the cores, the iron/aluminum ratio decreases continu-ously. In theory, individual crystals should anneal out these internal composition differences but in practice, cooling rates are too fast and diffusion too slow to permit internal homogenization on a micron scale. The silica content of ferrite is low, but the existence of a little silica in solid solution reduces the potential of the freezing melt to precipitate additional belite.
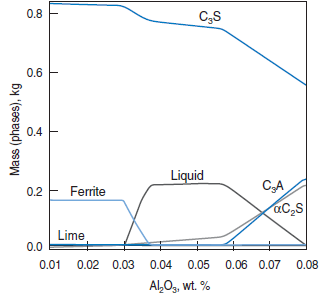
Figure 3.4.16. Equilibrium phases at 1350°C as a function of mass fraction of Al2O3 for com-position C = 0.69, S = 0.22, and (A + F) = 0.09. Note that in the range of high liquid contents, ferrite, and C3A are either absent or constitute a low mass fraction of the clinker.
The cumulative phase distributionarising from Scheil cooling of the example composition is shown in
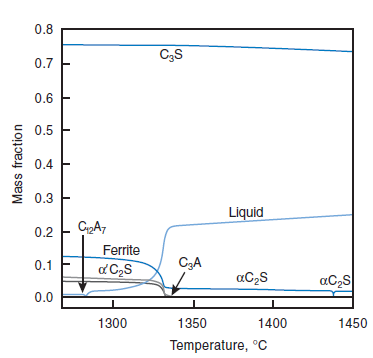
Figure 3.4.17. The accumulated mass of each of the indi-vidual phases formed during Scheil cooling of a clinker of overall composition C = 0.69, F = 0.036, A = 0.054, S = 0.22 from 1450°C, including the C2S and C3S that are unmelted at 1450°C. The mass of αC2S has been trans-ferred to α´C2S at 1438°C. A comparison with Figure 3.4.3 reveals important differences in the fraction of C3S at temperatures below 1340°C and the continuation of iquid below 1338°C. The proportions of ferrite, C2S,and C3A are also different.
Figure 3.4.17. Since belite exhibits polymorphism, changes in the amount and composition of belite also occur in the course of inversion: the remelting reaction and its
consequences have been described. Table 3.4.3 shows results of calculate phase compositions using different schemes for the example clinker. The Bogue calculation gives virtually identical results to a computer calculation, provided it is assumed that equilibrium is attained throughout freezing. This is proof of the essential soundness of the Bogue calculation. However,if equilibrium is not maintained – aprerequisite of the Bogue method –and if the basis of the calculation is altered to assume Scheil cooling, as appears to correspond to reality –calculated C3S increases sharply mainly at the expense of C2S, which decreases. The predicted small amount of C12A7 either escapes detection or persists as glass, or both. The main conclusion of the calculation, supported by experience, is that the phase composition
of cement departs from equilibrium during cooling, mainly by failure of previously-formed crystals to react with melt. This enhances the alite content relative to predictions made considering only equilibrium.With the advent of much more precise computational methods to link bulk composition with clinker mineralogy, cement makers should be able to reduce the variability inherent in presently available methods of controlling clinker mineralogy.
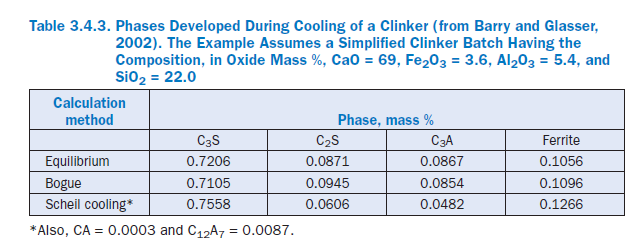
SUMMARY
The review demonstrates that advances in understanding the physical chemistry of clinkering enables better control of clinker mineralogy and reactivity. Many of the advances described here have yet to be fully exploited but, as manufacturers enter a new and challenging era of CO2 reduc-tion and environmental controls, further progress can be expected.
APPENDIX
Lime Saturation Factor
The lime saturation factor (LSF) calculates the maximum amount of CaO in the raw meal which can be combined into aluminates and silicates without excess of free lime. This condition is achieved when all silica is combined as C3S, so the (LSF) can be determined from the Bogue calcu-lation by setting C2S = 0. Alternatively, and assuming that ferrite has the C4AF composition, in weight % of oxides:

REFERENCES
Aldridge, L. P., “Accuracy and Precision of Phase Analysis in Portland Cement by Bogue, Microscopic and X-ray Diffraction Methods,” Cement and Concrete Research, Vol. 12, 1982,pages 381-398.
Barry, T. I., and Glasser, F. P., “Calculation of Portland Cement Clinkering Reactions,” Advances in Cement Research, Vol. 12, 2002, pages 19-28
.
Bhatty, J. I., Role of Minor Elements in Cement Manufacture and Use, RD109, Portland Cement Association, Skokie, Illinois, 1995, 40 pages.
Cahn, R. W., and Hassen, “Solidification,” Physical Metallurgy, Vol. 1, North Holland, Amsterdam, 1996, pages 670-844.
Campbell, D. H., Microscopical Examination and Interpretation of Portland Cement and Clinker, 2nd ed., SP030, Portland Cement Association, Skokie, Illinois, 1999.
Choi, G. S., and Glasser, F. P., “The Sulfur Cycle in Cement Kilns: Vapour Pressures and the Solid-Phase Stability of the Sulfate Phases,” Cement and Concrete Research, Vol. 18, 1988, pages 367-374.
Fletcher J. G., and Glasser, F. P., “Phase Relations in the System CaO-B2O3-SiO2,” Journal of Material Science, Vol. 28, 1993, pages 2677-2686.
Fukuda, K., “Recent Progress in Crystal Chemistry of Belite: Intracrystalline Microtextures Induced by Phase Transformations and Application of Remelting Reaction to Improvement of Hydration Reactivity,” Journal of Ceramic Society of Japan, Vol. 109, in English, 2001, pages 543-548
.
Fukuda, K., and I. Suketoshi, I., “Highly Reactive Remelted Belite,” Journal of American Ceramic Society, Vol. 82, 1999, pages 637-640.
Fukuda, K., et al “Characterization of Liquid Exsolved by Remelting Reaction of Belite,” Journal of American Ceramic Society, Vol. 84, 2001, pages 1155-1160.
Fukuda, K.; Takeda, A.; and Yoshida, H., “Remelting Reaction of Ca2SiO4 Solid Solution Confirmed in Ca2SiO4-Ca12Al14O33 Pseudobinary System,” Cement and Concrete Research, Vol. 31, 2001, pages 1185-1189.
Glasser, F. P., “The Burning of Portland Cement,” Chapter 5 in Lea’s Chemistry of Cement and Concrete, 4th ed., P.C. Hewelett (Editor), Edward Arnold, London, 1998, pages 195-240.
Glasser, F. P., and Marr, J., “Sulphates in Cement Clinkering: Immiscibility Between Sulfate and Oxide Melts at 1350°C,” Cement and Concrete Research, Vol. 10, 1980, pages 753-758.
Gutteridge, W. A., “Quantitative X-ray Powder Diffraction in the Study of Some Cementive Minerals,” Proceedings of the British Ceramic Society, Vol. 35, 1984, pages 11-23.
Han, K. S.; Gard, J. A.; and Glasser, F. P., “Compositions of Stable and Metastable C3A solid solu-tions Crystallized from Simulated Clinker Melts,” Cement and Concrete Research, Vol. 11, 1981, pages 79-84.
Hewlitt, P. C. (Ed.), Lea’s Chemistry of Cement and Concrete, Edward Arnold, London, 1998.
Insley, H., “Structural Characteristics of Some Constituents of Portland Cement Clinker,” Journal of Research and National Bureau of Standards, Vol. 17, R.P. 917, 1936, pages 353-361.
Kristmann, M., “Portland Cement Clinker: Mineralogical and Chemical Investigations, Part I. Microscopy, X-ray Fluorescence and X-ray Diffraction,” Cement and Concrete Research, Vol. 7, 1977, pages 649-658.
Kristmann, M., “Portland Cement Clinker: Mineralogical and Chemical Investigations. Part II. Electron Microprobe Analysis,” Cement and Concrete Research, Vol. 8, 1978, pages 93-102.
Kurdowski, W., “Role of Delayed Release of Sulphates from Clinker in DEF,” Cement and Concrete Research, Vol. 32, 2002, pages 401-408.
Maki, I., “Processing Conditions of Portland Cement Clinker as Viewed From the Fine Textures of the Constituent Minerals,” Ceramic Transactions, American Ceramic Society, Westerville Ohio, Vol. 40, 1994, pages 3-17.
Maki, I., et al “Formation of Belite Clusters from Quartz Grains in Portland Cement Clinker,” Cement and Concrete Research, Vol. 25, 1995, pages 835-840.
Mohan, K., and Glasser, F. P., “The Thermal Decomposition of Ca3SiO5 at Temperatures Below 1250°C Pure C3S and the Influence of Excess CaO or Ca2SiO4 Part I,” Cement and Concrete Research, Vol. 7, 1977, pages 1-8.
Mohan, K., and Glasser, F. P., “The Thermal Decomposition of Ca3SiO5 at Temperatures Below 1250°C Pure C3S and the Influence of Mg, Fe, Al, Na Oxides on the Decomposition Part II,” Cement and Concrete Research, Vol. 7, 1977, pages 269-0275.
Mohan, K., and Glasser, F. P., “The Thermal Decomposition of Ca3SiO5 at Temperatures Below 1250°C Pure C3S and the Influence of Water and Sulfate on the Decomposition Part III,” Cement and Concrete Research, Vol. 7, 1977, pages 379-383.
Norris, A., Computational Chemistry, John Wiley, Chichester, U.K., 1981.
Peray, K., The Rotary Cement Kiln, 2nd ed., Edward Arnold, London, 1986.
Pollitt, H. W. W., and Brown A.W., “The Distribution of Alkalis in Portland Cement Clinker,” 5th International Symposium on the Chemistry of Cement, Tokyo, Japan, Vol. 1, 1968, pages 252-261.
Saunders, N., and Miodownik, A. P., “CALPHAD, Calculation of Phase Diagrams: A Comprehensive Guide,” Pergamon Materials Series, Editor, Cahn, R.W., Vol. 1, 1998, Pergamon, Oxford, U.K.
Shame, E. G., and Glasser, F. P., “Synthesis and Properties of Stable Ca3SiO5 Solid Solutions Made between 1050°C and 1250°C,” British Ceramic Transaction and Journal, Vol. 86, 1987, pages 13-17.
Taylor, H. F. W., “Modification of the Bogue Calculation,” Advances in Cement Research, Vol. 2, 1989, pages 73-77.
Terrier, P., “Contribution of Analysis by Means of an Electron Microprobe to the Cement Chemistry,” 5th International Symposium on the Chemistry of Cement, Tokyo, Japan, Vol. 2, 1968, pages 278-287.
Yamaguchi, G., and Ono, U., “Microscopic Studies on the Texture of Belite in Portland Cement Clinker,” 16th General Meeting of the Cement Association of Japan Reviews, 1962, pages 32-34.
Yamaguchi, G., and Takagi, S., “The Analysis of Portland Cement Clinker,” 5th International Symposium on the Chemistry of Cement, Tokyo, Japan, Vol. 1, 1968, pages 181-225.